DeRIP2 Command Line Interface
Basic usage
For aligned sequences in 'mintest.fa':
- Any column with >= 70% gap positions will not be corrected and a gap inserted in corrected sequence.
- Bases in column must be >= 80% C/T or G/A
- At least 50% bases in a column must be in RIP dinucleotide context (C/T as CpA / TpA) for correction.
- Default: Inherit all remaining uncorrected positions from the least RIP'd sequence.
- Mask all substrate and product motifs from corrected columns as ambiguous bases (i.e. CpA to TpA --> YpA)
Basic usage with masking
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--mask \
-d results \
--prefix derip_output
Output:
results/derip_output.fasta- Corrected sequenceresults/derip_output_alignment.fasta- Alignment with masked correctionsresults/derip_output_masked_alignment.fasta- Alignment with masked corrections
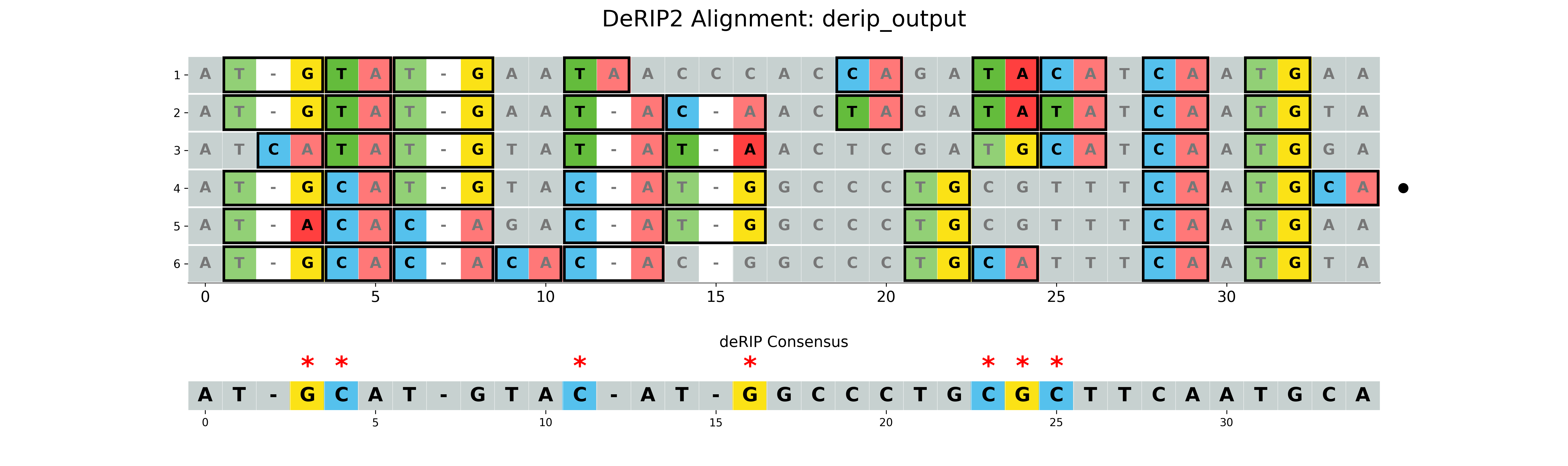
With vizualization
The --plot option will create a visualization of the alignment with RIP markup. The --plot-rip-type option can be used to specify the type of RIP events to be displayed in the alignment visualization product, substrate, or both.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--plot \
--plot-rip-type both \
-d results \
--prefix derip_output
Output:
results/derip_output.fasta- Corrected sequenceresults/derip_output_masked_alignment.fasta- Alignment with masked correctionsresults/derip_output_visualization.png- Visualization of the alignment with RIP markup
Using maximum GC content for filling
By default uncorrected positions in the output sequence are filled from the sequence with the lowest RIP count. If the --fill-max-gc option is set, remaining positions are filled from the sequence with the highest G/C content sequence instead.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--fill-max-gc \
-d results \
--prefix derip_gc_filled
Alternatively, the --fill-index option can be used to force selection of alignment row to fill uncorrected positions from by row index number (indexed from 0). Note: This will override the --fill-max-gc option.
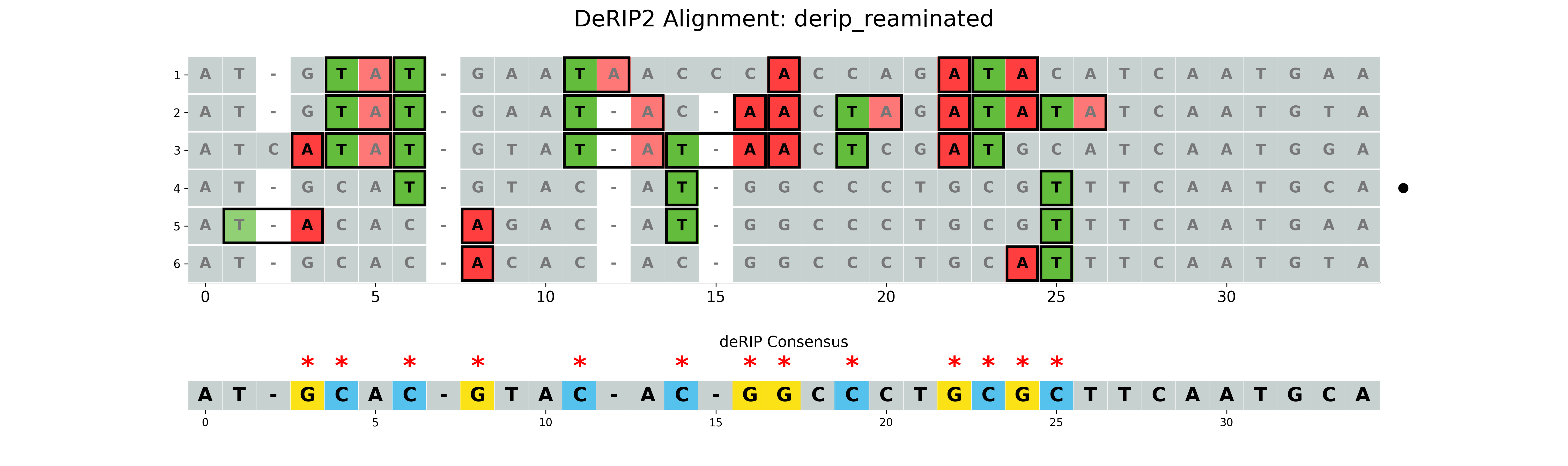
Correcting all deamination events
If the --reaminate option is set, all deamination events will be corrected, regardless of RIP context.
--plot-rip-type product is used to highlight the product of RIP events in the visualization.
Non-RIP deamination events are also highlighted.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--reaminate \
-d results \
--plot \
--plot-rip-type product \
--prefix derip_reaminated
Output:
results/derip_reaminated.fasta- Corrected sequence using highest GC content sequence for fillingresults/derip_reaminated_alignment.fasta- Alignment with corrected sequence appendedresults/derip_reaminated_vizualization.png- Visualization of the alignment with RIP markup
Standard Options
--version Show the version and exit.
-i, --input TEXT Multiple sequence alignment. [required]
-g, --max-gaps FLOAT Maximum proportion of gapped positions in
column to be tolerated before forcing a gap
in final deRIP sequence. [default: 0.7]
-a, --reaminate Correct all deamination events independent
of RIP context.
--max-snp-noise FLOAT Maximum proportion of conflicting SNPs
permitted before excluding column from
RIP/deamination assessment. i.e. By default
a column with >= 0.5 'C/T' bases will have
'TpA' positions logged as RIP events.
[default: 0.5]
--min-rip-like FLOAT Minimum proportion of deamination events in
RIP context (5' CpA 3' --> 5' TpA 3')
required for column to deRIP'd in final
sequence. Note: If 'reaminate' option is set
all deamination events will be corrected.
[default: 0.1]
--fill-max-gc By default uncorrected positions in the
output sequence are filled from the sequence
with the lowest RIP count. If this option is
set remaining positions are filled from the
sequence with the highest G/C content.
--fill-index INTEGER Force selection of alignment row to fill
uncorrected positions from by row index
number (indexed from 0). Note: Will override
'--fill-max-gc' option.
--mask Mask corrected positions in alignment with
degenerate IUPAC codes.
--no-append If set, do not append deRIP'd sequence to
output alignment.
-d, --out-dir TEXT Directory for deRIP'd sequence files to be
written to.
-p, --prefix TEXT Prefix for output files. Output files will
be named prefix.fasta,
prefix_alignment.fasta, etc. [default:
deRIPseq]
--plot Create a visualization of the alignment with
RIP markup.
--plot-rip-type [both|product|substrate]
Specify the type of RIP events to be
displayed in the alignment visualization.
[default: both]
--loglevel [DEBUG|INFO|WARNING|ERROR|CRITICAL]
Set logging level. [default: INFO]
--logfile TEXT Log file path.
-h, --help Show this message and exit.
Mutation spectra (derip2-spectra)
derip2-spectra builds trinucleotide-context SBS-96 and SBS-192 mutation spectra
from an alignment. See the Mutation Spectra tutorial
for a full walkthrough; the essentials are below.
Baseline (no tree, no external tools)
Writes family.SBS96.txt, family.SBS192.txt (SigProfiler-compliant matrices),
the spectrum/strand-asymmetry/homoplasy plots, and family_events.tsv.
By default the baseline reconstructs the deRIP consensus internally and calls every sequence against it.
Reusing a precomputed ancestral reference
If you have already run derip2 and appended the deRIP'd consensus to your
alignment, derip2-spectra will reuse it instead of recomputing. Any row whose
id matches --reference-tag (default deRIPseq) is used as the ancestral
reference and excluded from the counted sequences (a message is logged when
this happens):
# family_with_deRIPseq.fasta already contains a "deRIPseq" row
derip2-spectra -i family_with_deRIPseq.fasta -d results -p family
# Point at a differently-named reference row
derip2-spectra -i family.fasta --reference-tag MyAncestor -d results -p family
Alternatively, supply a separate hypothetical ancestor as a single-sequence
FASTA with --ancestor. It must be the same length as the alignment (this is
validated up front) and takes precedence over any in-alignment reference row:
Input must be unambiguous DNA. Alignments may only contain
A/C/G/T/-(upper or lower case; soft-masking is normalised). Degenerate IUPAC characters (N,R,Y, …) are rejected with an error naming the offending character and its location, rather than being silently treated as gaps.
Phylogenetic (IQ-TREE ancestral reconstruction)
Requires IQ-TREE (iqtree3/iqtree2/iqtree) on PATH.
# Infer the tree and reconstruct ancestors automatically
derip2-spectra -i family.fasta --method phylo -d results -p family
Supplying a precalculated phylogeny
Pass any Newick tree with --tree; IQ-TREE fixes that topology and recomputes the
model, branch lengths and ancestral states from the alignment.
iqtree3 -s family.fasta -m MFP -B 1000 -T AUTO --prefix family_tree
derip2-spectra -i family.fasta --method phylo --tree family_tree.treefile \
-d results -p family
Recommended: topology from a RIP-masked alignment
Infer the topology from a RIP-masked alignment (so convergent RIP does not distort it), then reconstruct ancestral states for the unmasked sequences on that same topology:
derip2 -i family.fasta --mask --no-append -d results -p family
iqtree3 -s results/family_masked_alignment.fasta -m MFP -B 1000 -T AUTO \
--prefix results/family_masked
derip2-spectra -i family.fasta --method phylo --tree results/family_masked.treefile \
-d results -p family_spectrum
Per-group spectra (species or user-defined sets)
Pass a two-column (name, group) file with --groups to report one spectrum per
group. Works for both methods and tolerates IQ-TREE name reformatting:
derip2-spectra -i family.fasta --groups groups.tsv -d results -p family
derip2-spectra -i family.fasta --method phylo --groups groups.tsv -d results -p family
Run derip2-spectra --help for the full option list (--sbs, --rooting,
--outgroup, --partition-by, --groups, --min-prob, --root-sensitivity,
--threads, …).